فیلوژنتیک
فیلوژنتیک یا تبارزایش (به انگلیسی: Phylogenetics) شاخهای در علم زیستشناسی میباشد که به بررسی ارتباط تکاملی گروههای مختلف جاندران نظیر گونهها یا جمعیتها میپردازد، که از دادههای توالییابی مولکولی[پانویس 1] و ماتریسهای دادههای ریختشناسی[پانویس 2] بهدست میآید.
واژه فیلوژنتیک از ریشه یونانی فیل (φυλή/φῦλον) به معنی «تبار»، و ژنتیکوس (γενετικός) به معنی «مربوط به زایش» یا زایشی گرفته شدهاست. آرایهشناسی، طبقهبندی، شناسایی و نامگذاری جانداران بسیار از فیلوژنتیک کمک گرفتهاند، ولی همچنان از نظر روششناسی و منطقی از آن مجزا هستند.[1]
تبارزایش با علم سامانهشناسی فیلوژنتیک[پانویس 3] (اکثراً کلادگرایی یا شاخهبندی خوانده میشود) وجوه اشتراک دارد، که در آن تنها درختان فیلوژنتیک برای محدود کردن آرایه و نمایش گروههای دودمانی وابسته استفاده میشوند.[2] در سامانهشناسی زیستی، تحلیل فیلوژنتیک به ابزاری ضروری برای پژوهش روی درخت فرگشتی حیات مبدل شدهاست.
ساخت درخت تبارزایشی
تکامل را میتوان یک روند انشعاب دانست، روندی که به موجب آن جمعیتهایی از موجودات زنده با گذر زمان تغییر میکنند و در اثر تغییرات به شاخههای جداگانهای گونهزایی میکنند، با هم درمیآمیزند (ادغام میشوند)، یا به واسطه انقراض از میان میروند. این روند را میتوان توسط یک درخت فیلوژنتیکی مجسم کرد.
مشکل اساسی در فیلوژنتیک این است که اطلاعات ژنتیکی فقط برای آرایههای امروزی موجد هستند، و سوابق فسیلی حاوی اطلاعات کم و شاخصههای ریختشناختی مبهمتری هستند. یک درخت فیلوژنتیک نشان دهنده فرضیهای است دربارهٔ روندی که طبق آن رویدادهای تکاملی فرض میشود که اتفاق افتادهاند.
کلاد بندی روش مرسوم برای استنباط درخت فیلوژنتیک است. معمولترین روشهای مورد استفاده برای استنباط فیلوژنی شامل پارسیمونی (یا کمترین فرضیات)، درستنمایی حداکثری، و استنتاج بیزی مبتنی بر MCMC هستند. فنتیک، روشی محبوب در اواسط قرن بیستم که در حال حاضر تا حد زیادی منسوخ شده، از روش ماتریس فاصله استفاده میکند تا درختی ایجاد کند مبتنی بر تشابهات شکلی، که تصور میشود تقریب خوبی از روابط فیلوژنتیکی باشد. همه این روشها وابسته به مدلهای ریاضی ضمنی یا صریحی هستند که تکامل شاخصههای مشاهده شده در گونههای مورد مطالعه را توضیح میدهند. این مدلها معمولاً بر اساس دادههای مولکولی ساخته میشوند، و شاخصهها در این نوع مدلها نوکلئوتیدها یا رشتههای اسید آمینه برخط شده[پانویس 4] هستند.
گروهبندی جانداران
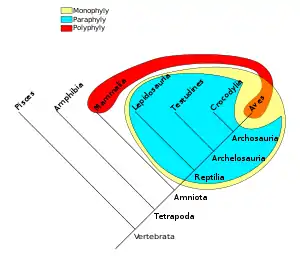
گروههای فیلوژنتیک یا آرایهها میتوانند گروه فراگیر monophyletic، گروه نافراگیر paraphyletic یا گروه چندنیا polyphyletic باشند. به عنوان نمونه شواهد نشان میدهد که همه پرندگان و خزندگان از یک نیای مشترک پدید آمدهاند بنابراین با قرار دادن آنها در یک دسته گروهی تشکیل میشود که هم نیای اصلی و هم همه نوادگان او را دربر میگیرد و بنابراین یک گروه فراگیر (زرد در شکل پایین) است. از آنجا که پرندگان تفاوتی عمده با خزندگان پیدا کردهاند زمانی که آنها را از دسته خزندگان برداشته و حذف کنیم یک گروه نافراگیر (آبی متمایل به سبز در شکل) باقی میماند. یک نوع دستهبندی دیگر در نظر گرفتن گروهی به نام جانوران خونگرم است که شامل تنها پستانداران و پرندگان میشود (قرمز / نارنجی در شکل) به این دسته، گروه چندنیا گفته میشود زیرا اعضای آن بهتازگی نیای مشترک نداشتهاند.
تبارزایش مولکولی
ارتباط تکاملی بین موجودات به وسیله درخت فیلوژنتیک نمایش داده میشود. از آنجا که تکامل در طول دورههای زمانی طولانی که بهطور مستقیم قابل مشاهده نیست اتفاق میافتد، زیستشناسان مجبورند فیلوژنیها را با استنباط روابط تکاملی میان جانداران امروزی بازسازی کنند. سنگوارهها میتوانند به بازسازی فیلوژنیها کمک کنند؛ اما، رکوردهای فسیلی اغلب بیش از حد کمیاب هستند و کمک خوبی نیستند؛ بنابراین، زیستشناسان معمولاً به تجزیه و تحلیل موجودات امروزی برای شناسایی روابط تکاملی آنها محدود هستند. روابط تبارزایی در گذشته از روی ویژگیهای فنوتیپ، که اغلب شاخصههای کالبدشناختی میباشند، بازسازی میشدند. امروزه، دادههای مولکولی، شامل پروتئین و رشتههای دیانای، برای ساخت درختهای فیلوژنتیک استفاده میشوند.[3]
هدف کلی از پروژهٔ ساخت درخت زندگی (AToL) که بنیاد ملی علوم آمریکا آن را راه انداخته، آنست که روابط تکاملی گروههای زیادی از موجودات در طول تاریخ حیات را کشف کند. این پژوهشها اغلب شامل تیمهای بزرگی میشود که در موسسات و رشتههای متنوع کار میکنند.
نظریه رشد و نمو ارنست هکل
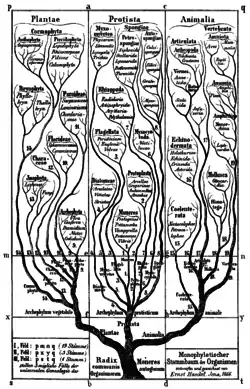
در اواخر قرن ۱۹ میلادی نظریه تکرار رشد و نمو هکل، یا قانون زیستی بهطور گسترده پذیرفته شد. این نظریه اغلب به عنوان "ontogeny recapitulates phylogeny" شناخته میشود. یعنی پیشرفت و تکامل یک موجود دقیقاً بازتاب پیشرفت وتکامل ان گونهاست.[4] (ارنست هکل را ببینید)
تبادل ژن
بهطور معمول ارگانیسمها به دو طریق میتوانند ژنها را به ارث ببرند: انتقال افقی ژن و انتقال عمودی ژن. انتقال افقی ژن شامل انتقال ژن از والد به فرزندان است. انتقال غمودی ژن یا انتقال جانبی ژن وقتی اتفاق میافتد که ژن بین دو ارگانیسم بدون ارتباط جهش کند. این یک پدیده معمولاً در تک سلولیها است. یک مثال خوب از این نوع ساخت آنتیبادیهای مقاوم به عنوان نتیجه تبادل ژن بین بعضی از باکتریها و MDR است.
انتقال عمودی ژن، امر مشخصسازی ساختار نژادی یک گونه را دشوار میکند؛ و بر حسب اینکه چه ژنی در ساخت درخت تکاملی به کار گرفته شده باشد، ناهماهنگیهایی در ساختار ژنی در بین گونههای خاص از موجودات گزارش شدهاست.
کارل وز، با تئوری سه بعدی حیات (eubacteria, archaea and eukaryota) شناخته میشود. این نظریه بر پایه کشف خود او که ژنهایی که RNAهای داخل ریبوزوم (rRNA) را دیکد میکنند از قدیم داخل ارگانیسمها بوده و در کل شکل حیات توزیع شدهاند؛ و انتقال عمودی ژن یا بر روی آنها تأثیر نگذاشته یا کمترین تأثیر را گذاشتهاست. بنابراین میتوان از rRNA به عنوان یک ساعت مولکولی برای تشکیل ساختار ژنی استفاده کرد. این بهطور خاص برای ساختارشناسی میکرو جانداران مفید است.
نمونهگیری آرایه و علامت تبارزایشی
به علت توسعه تکنیکهای پیشرفته استخراج توالی در بایولوژی مولکولی امکان جمعآوری مقدار زیادی اطلاعات جهت استنتاج فرضیههای فیلوژنی فراهم شدهاست. به عنوان مثال پیدا کردن مقالاتی در مورد میتوکندری (با حدود ۱۶۰۰۰ نوکلئوتید در اکثر حیوانات) که بر پایه ماتریس کاراکتر هستند سادهاست. به هر حال افزایش taxaها خیلی اهمیت دارد زیرا افزایش taxaها به معنی افزایش دقت و در نتیجه ایجاد درخت فیلوژنی مقاوم تر است. در فیلوژنتیک ما انتظار دنبالههای نسبتاً طولانی را داریم و این یک دلیل محکم برای این است که در فیلوژنتیک، دادههای بدست آمده از سنگوارهها را هر جا که ممکن باشد، استفاده میکنیم. البته دادههای تبارزایشی فسیلها بیشتر شامل اطلاعات ریختشناسی است تا اطلاعات مربوط به DNA.
Derrick Zwickl و David Hillis با استفاده از شبیهسازی نشان دادند، افزایش نمونههای آرایه (زیستشناسی) در استنتاج فیلوژنتیک تأثیر مثبتی دارد و دقت آنالیز فیلوژنتیک را افزایش میدهد.[5]
عامل دیگری که در دقت درخت ساخته شده مؤثر است این است که آیا دادههای آنالیز شده، شاما اطلاعات سودمند فیلوژنتیک بودهاند یا نه. این عامل به عنوان یک ضابطه عادی برای تشخیص اینکه دو ارگانیشم شبیه به هم آیا دنباله فیلوژنتیک مشابه به هم نیز دارند، استفاده میشود.[6]
در نهایت هیچ راهی وجود ندارد که دقت یک نظریه خاص فیلوژنتیک را بررسی کند. مگر اینکه ارتباط دقیق آرایه (زیستشناسی)ها با استفاده از آزمایش بررسی شده باشد. بهترین نتیجه برای یک دستهبندی تجربی، میتواند یک درخت با انشعابهایی باشد که برای شواهد موجود خوب عمل میکند.
اهمیت دادههای نداشته
حالت عادی اگر داده بیشتری هنگام ساخت درخت فیلوژنی داشته باشیم، نتیجتاً درخت با دقت و ارتباط بالا خواهیم داشت. زیان دادههای گم شده کمتر از داشتن داده کم نیست. اگر چه موقعی که دادههای گم شده مربوط به تعداد کمی از taxaها باشد زیان کمتر است. متمرکز کردن دادههای گم شده در بین تعداد کمی از taxaها باعث تولید درخت مقاوم تر میشود.[7]
نقش سنگوارهها
به دلیل اینکه بسیاری از شاخههای ریختشناسی شامل رویانشناسی یا بافتشناسی است، که نمیتوانند سنگواره شوند و تفسیر قسیل بسیار گیجکننده تر از رده آرایه (زیستشناسی)های زندهاست، در برخی موارد بسیار مشکل است که دادههای فسیلی را برای فیلوژنتیک هماهنگ کرد. با وجود این محودودیتها اهمیت فسیل غیرقابل انکار است. زیرا آنها میتوانند اطلاعات مهمی در مورد فضاهای اسپارس در درختان فراهم کنند و انشعابات طولانی را بشکنند و به عنوان راس میانی قرار گیرند. بنابراین فسیلها به همان اندازه که taxaهای جدید اهمیت دارند، مهم هستند.[8] فیلوژنتیک مولکولی میتواند نرخ تنوع ارگانیسمها را مشخص کند. اما برای فهمیدن دنباله ایجاد و انقراض گونهها و شناخت الگوها در تنوع ارگانیسمها، دادههای فسیلی باید استفاده شوند.[7] تکنیکهای مولکولی یک نرخ ثابت تنوع فرض میکنند، که به ندرت با واقعیت سازگار است، اما در مورد دادههای فسیلی اینگونه نیست.
سبکوسنگینکردن هوموپلازی
بعضی از کاراکترهای مشخص با احتمال زیاد به همگرایی منجر میشوند. منطقا این کتراکترها باسد وزن کمتری در ساخت درخت داشته باشند. متأسفانه تنها راه مشخص کردن همگرایی ساخت درخت است.[9] وزن دار کردن کاراکترهای متشابه جانوری، به درخت با ساختار بهتر منجر میشود.[9]
جستارهای وابسته
یادداشت
- Molecular Sequencing Data
- Morphological Data Matrices
- Phylogenetic nomenclature (PN) or phylogenetic
- Aligned
منابع
- A.W.F. Edwards, [L.L. Cavalli-Sforza
Phylogenetics is that branch of life science,which deals with the study of evolutionary relation among various groups of organisms,through molecular sequencing data. (1964). Systematics Assoc. Publ. No. 6: Phenetic and Phylogenetic Classification, ed. Reconstruction of evolutionary trees. pp. 67–76. line feed character in
|author=at position 37 (help) - Speer, Vrian (1998). "UCMP Glossary: Phylogenetics". UC Berkeley. Archived from the original on 29 September 2017. Retrieved 2008-03-22.
- Pierce, Benjamin A. (2007-12-17). Genetics: A conceptual Approach (3rd ed.). W. H. Freeman. ISBN 978-0-7167-7928-5.
- Michael W. Hart, and Richard K. Grosberg, "Caterpillars did not evolve from onychophorans by hybridogenesis", Proceedings of the National Academy of the Sciences, 30 October 2009 (doi: 10.1073/pnas.0910229106)
- Zwickl DJ, Hillis DM (2002). "Increased taxon sampling greatly reduces phylogenetic error". Systematic Biology. 51 (4): 588–598. doi:10.1080/10635150290102339. PMID 12228001.
- Blomberg SP, Garland T Jr, Ives AR (2003). "Testing for phylogenetic signal in comparative data: behavioral traits are more labile". Evolution. 57 (4): 717–745. PMID 12778543. PDF بایگانیشده در ۱۶ ژوئن ۲۰۱۰ توسط Wayback Machine
- Quental, Tiago B.; Marshall, Charles R. (2010). "Diversity dynamics: molecular phylogenies need the fossil record". Trends in Ecology & Evolution. 25 (8): 434–441. doi:10.1016/j.tree.2010.05.002. ISSN 0169-5347.
- Cobbett, Andrea; Wilkinson, Mark; Wills, Matthew A; Sullivan, Jack (2007). "Fossils Impact as Hard as Living Taxa in Parsimony Analyses of Morphology". Systematic Biology. 56 (5): 753–766. doi:10.1080/10635150701627296. ISSN 1076-836X.
- Goloboff, Pablo A.; Carpenter, James M.; Arias, J. Salvador; Esquivel, Daniel Rafael Miranda (2008). "Weighting against homoplasy improves phylogenetic analysis of morphological data sets". Cladistics. 24 (5): 758–773. doi:10.1111/j.1096-0031.2008.00209.x. ISSN 0748-3007.
- مشارکتکنندگان ویکیپدیا. «Phylogenetics». در دانشنامهٔ ویکیپدیای انگلیسی، بازبینیشده در ۲۷ نوامبر ۲۰۱۰.
پیوند به بیرون
- دانشنامه درخت حیات
- درخت زندگی اینترکتیو
- نمایشگاه فیلوژنی
- انجمن ویلی هنیگ
- SplitsTree، نرمافزار محاسبات فیلوژنتیک
- Dendroscope، نرمافزار نمایش درخت فیلوژنتیک
- سیستم پیمانهای برای تحلیل فرگشتی Mesquite
- Geneious Pro نرمافزار فیلوژنتیک چندکاره
- NCBI - سامانهشناسی و فیلوژنتیک ملکولی
- PhylomeDB - پایگاه داده عمومی فیلوژنی ژن
| مباحث مرتبط |
| |
|---|---|---|
| مفاهیم پایهای |
| |
| روشهای استنباط |
| |
| موضوعات جاری |
| |
| خصایص گروهی |
| |
| انواع گروه | ||
| نامگذاری |
| |
| ||
| ||
| فرگشت |
|  |
| ژنتیک جمعیت | ||
| تکوین |
| |
| آرایه |
| |
| عضو |
| |
| فرآیند |
| |
| تمپوها و مدها |
| |
| گونهزایی |
| |
| تاریخچه اندیشه فرگشتی |
| |
| فلسفه |
| |
| مرتبط |
| |
| ||
