پریون
پریان یا پریون (![]() i/ˈpriːɒn/[1]) انواعی از پروتئین است که میتواند به شکلهای متنوعی تا شود. آنها معمولاً بیماریزا هستند و در گروههای مختلف موجودات ایجاد بیماری میکنند.
هیچ اسید نوکلئیک قابل شناسایی ندارند. آنها در برابر فرمالدئید و گرما و اشعه ماورابنفش که ویروسها را غیرفعال میکنند کاملاً مقاوم هستند. بیماریهای پریونی را که انسفالوپاتی اسفنجی شکل مسری مینامند شامل بیماری اسکراپی در گوسفندان و بیماری جنون گاوی در گاوها.
i/ˈpriːɒn/[1]) انواعی از پروتئین است که میتواند به شکلهای متنوعی تا شود. آنها معمولاً بیماریزا هستند و در گروههای مختلف موجودات ایجاد بیماری میکنند.
هیچ اسید نوکلئیک قابل شناسایی ندارند. آنها در برابر فرمالدئید و گرما و اشعه ماورابنفش که ویروسها را غیرفعال میکنند کاملاً مقاوم هستند. بیماریهای پریونی را که انسفالوپاتی اسفنجی شکل مسری مینامند شامل بیماری اسکراپی در گوسفندان و بیماری جنون گاوی در گاوها.
| Prion diseases | |
|---|---|
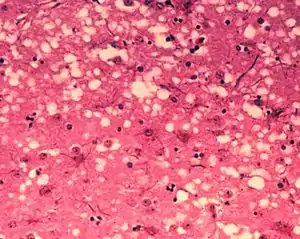 | |
| Microscopic "holes" are characteristic in prion-affected tissue sections, causing the tissue to develop a "spongy" architecture. This causes deterioration of that "spongy" tissue in the brain. | |
| طبقهبندی و منابع بیرونی | |
| آیسیدی-۱۰ | A81 |
| آیسیدی-9-CM | 046 |
پریون یکی از پروتئینهایی است که بهطور عادی در سلولهای عصبی بیان میشود. در واقع شکل سلولی پروتئین پریون به وسیله کروموزوم میزبان رمزدهی میشود.
پریون سلولی، نوعی سیالوگلیکوپروتئین، با وزن مولکولی ۳۵–۳۳ هزار است که در ساختمان دومین آن، مارپیچهای آلفا به تعداد زیاد وجود دارد. این پروتئین نسبت به اثر پروتئازها حساس بوده، در گندزداها محلول میباشد. تنها جزء شناخته شده از پریون، یک نوع مشابه اما غیرطبیعی از این پروتئین است که با مسری بودن بیماری مرتبط است. این پروتئین غیرطبیعی دارای توالی اسیدآمینههای مشابه با پریون سلولی است. اما از لحاظ فیزیکی با دارا بودن صفحههای بتای فراوان، غیر محلول بودن در گند زداها، تمایل داشتن به تجمع و مقاومت نسبی در برابر پروتئولیز، با پروتئین طبیعی متفاوت است.[2]
PrPCنوعی پروتئین عادی است که در غشای سلولی سلولهاست، که در انسان 209آمینواسید،یک پیوند دی-سولفیدی دارد.از لحاظی مکانی انواعی دارد:یک نوع به سطح سلول با گلیکولیپید ها چسبیده و دیگر نوع، از نوع پروتئین انتقالی تمام عرض غشایی است.این پروتئین ته نشین نمیشود، یعنی با روشهای سانتریفیوژ ،قابلیت جداسازی ندارد. این پروتئین با یون مس دومثبت(+Cu2)، پیوند سختی برقرار میکند.هرچند عملکرد آن هنوز در دست تحقیق است، اما گزارش شده که نقش مهمی در چسبیدگی سلول و سلول به هم،و سیگنال و علامت دهی درون سلولی –و در نتیجه در ارتباط سلول ها در مغز- داشته باشد.این پروتئین ها در سراسر بدن افراد حتی سالم هم یافت میشوند.
PrPresدر واقع ایزوفرم پریون غیربیماریزا است که دربرابر پروتئاز مقاوم است.
که طبق ساختار (میس-فولد شده یا به اشتباه تاخورده)به این آنزیم مقاوم است که طی فرایندی
که پریون غیر بیماریزا به سرعت و بدون دقت به این پروتئین تبدیل شده است.اما علت اینکه نام
این پروتئین با پروتئین بیماریزا فرق میکند،اینست که این پروتئین به اشتباه تا خورده لزوماً عفونی نیست و سبب ایجاد بیماری بافت عصبی اسفنجی شکل نمیشود.
تفاوت بین فرم بیماریزا و غیر بیماریزا در ساختمان سوم این دو ذره است. یعنی در پریون عادی α-Helix42% 3% β-sheets است.
اما در پریون بیماریزا 43% β-sheets و 3% α-helix وجود دارد.که این تغییرات ساختاری تغییرات گسترده بیوشیمایی در بر دارد و سبب مقاومت فرم بیماری زا می شود. فرم PrPcبرابر پروتئاز حساس بوده و در مواد پاک کننده محلول است و بیشتر ساختار آن را زنجیر آلفا تشکیل می دهد. اما فرم PrPscبیشتر دارای زنجیره بتا می باشد که در مواد پاک کننده نامحلول بوده و دارای مقاومت نسبی در مقابل پروتئولیز می باشد. شایان ذکر است به دلیل نداشتن اسید نوکلئیک در ساختار پریون این ذرات به نوکلئاز و UV 240nmمقاوم هستند. پریونها میتواند مجتمع شده و پلاکهای پروتئینی را در سلولهای عصبی ایجاد کند. این پلاکها باعث انسفالوپاتیهای اسفنجی شکل در مغز میشوند.
ساختمان پریونها فقط از پروتئین ساخته شده و فاقد اسید نوکلئیک است. بیماریهای ناشی از پریون در انسان به علت اینکه به صورت بیماریهای ژنتیکی و عفونی بروز میکند کاملاً اختصاصی هستند.
فرایند تولید پریون های (ری-فولد شده یا باز تاخورده) که بیماریزا و عفونی هستند میتوانند توسط پروتئین های چپرونی مثل Hsp104pصورت گیرد. این پریونها میروند و با برخورد تغیر و تبدیل دیگر پروتئین ها به پریون،واکنش زنجیروار تولید بسیار زیادی پریون را صورت میدهد.همه انواع پریون ها سبب القا و تولید تا خوردگی به شکل نشاسته میکنند،بگونه ای که پریون ها با صفحات بتای فراوان روی هم انباشته و پلیمریزه میشوند که نوعی فیبریل هستند که پریون به انتهای آنها اضافه میشود،این فرآید بارها تکرا میشود تا یک فیبریل با دو انتها بشکند و به دو فیبریل با چهار انتها تبدیل شود.این تکثیر به صورت رشد نمایی(نمودار نمایی) ادامه میابد که به رشد خطی پریونها و شکسته شدن این یبریل مرتبط است{.در نظر بگیرید که تکثیر پریون ها به وجود پروتئین های به طور طبیعی تا خورده وابسته است؛یعنی اگر در جانداری پروتئین عادی پریون بیان نشود، امکان انتقال بیماری نیست،-هماهنطور که در مثال های بعدی توضیح خواهیم داد-.
فرضیه سمیت فلزات سنگین: گزارشات اخیر حاکی از آنست که عدم وجود هومئوستازی و تعادل فلزات سنگین در بدن،یکی از مهم ترین علل تولید پریون بیماریزاست که سبب مسمومیت نورون میشود.البته با توجه به اطلاعات موجود، توضیح مکانیسم این فرایند دشوار است.در فرضیه،نقش عملکردی برای پریون بیماریزا در متابولیسم فلز، و کمبود این عملکرد بخاطر انباشته شدن بیماری های پریونی بعنوان علت عدم تعادل فلزات در مغز،فرض شده.شواهد دیگری نشان میدهد که عملکرد سمی پریون بیماریزا بخاطر تجزیه پریونهای عادی متصل به فلزات، و همراه با انباشته شدن اینها؛سبب تولید کمپلکس های پریون های بیماری زای فعال ریدُکس(اکسایشی کاهشی)میشود.آسیب شناسی دلالت دارد براینکه تعمل پریون عادی با فلز،شامل آسیب اکسیداتیو فلز کمپلکس، و طبق برخی مثال ها تبدیل پریون عادی به پریون بیماریزاست.
تشخیص
شایان ذکر است که ضد پریونها در بدن پاسخ ایمنیمصونیتزا ایجاد نمیشود، بنابراین تشخیص آلودگی یا بیماری با بررسی پادتنها غیرممکن است. تنها راه شناسایی عامل بیماری در بافت آسیبدیده میباشد؛ بنابراین گرچه بهترین راه تشخیص بیماریهای پریونی علایم بالینی میباشد ولی برای تشخیص قطعی بایستی بافت یا مایع مغزینخاعی مورد آزمایشهای پیشرفتهای قرار گیرد.
به تازگی مشخص شدهاست که پروتئینهایی مشابه پریونها به نام CPEB، میتوانند در تشکیل حافطه در مغز نقش داشته باشند. این پروتئینها در اثر سیگنالهای الکتریکی که از پیامهای عصبی میرسند تغییر فرم داده و به شکل پریونها در میآیند.
بیماریهای پریونی
تمام بیماریهای پریونی شناخته شده در مجموع انسفالوپاتیهای اسفنجی شکل قابل انتقال (TSEs) نامیده میشوند که غیرقابل درمان و کشندهاند.[3] گونههای زیادی از پستانداران به بیماریهای پریونی مبتلا میشوند، بهطوریکه پروتئین پریون (PrP) در تمام پستانداران خیلی به هم شبیه است.[4] با توجه به اختلافات کم بین PrP گونههای مختلف، انتقال بیماری پریونی از یک گونه به دیگری غیرمعمول است. بااینحال بیماری پریونی انسانی کورتز فلدت جاکوب به وسیلهٔ پریونی که بهطور معمول گاوها را آلوده میکند، ایجاد میشود. این پریون موجب ایجاد آنسفالوپاتی اسفنجی شکل گاوی میشود و از طریق گوشت آلوده به انسان منتقل میشود.[5]
جستارهای وابسته
منابع
| در ویکیانبار پروندههایی دربارهٔ پریون موجود است. |
- "Prion". فرهنگ انگلیسی آکسفورد. انتشارات دانشگاه آکسفورد. 2nd ed. 1989.
- Jawetz Melnick&Adelbergs Medical Microbiology 26/E (Jawetz, Melnick, & Adelberg's Medical Microbiology
- ^ Gilch S, Winklhofer KF, Groschup MH, et al. (August 2001). "Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease". The EMBO Journal 20 (15): 3957–66. doi:10.1093/emboj/20.15.3957. PMC 149175. PMID 11483499.
- ^ Collinge J (2001). "Prion diseases of humans and animals: their causes and molecular basis". Annual Review of Neuroscience 24: 519–50. doi:10.1146/annurev.neuro.24.1.519. PMID 11283320. Retrieved 2010-02-28.
- ^ Ironside JW (March 2006). "Variant Creutzfeldt-Jakob disease: risk of transmission by blood transfusion and blood therapies". Haemophilia: the Official Journal of the World Federation of Hemophilia 12 Suppl 1: 8–15; discussion 26–8. doi:10.1111/j.1365-2516.2006.01195.x. PMID 16445812. Retrieved 2010-02-28.
- Wikipedia contributors, "Prion," Wikipedia, The Free Encyclopedia, https://en.wikipedia.org/w/index.php?title=Prion&oldid=679144825 (accessed September 9, 2015).
- Si, K. et al. A neuronal isoform of CPEB regulates local protein synthesis and stablizes synapse-specific long-term facilitation in Aplysia. Cell, ۱۱۵، ۸۷۹–۸۹۱، (۲۰۰۳).
- Si, K. , Lindquist, S. & Kandel, E.R. A neuronal isoform of the Aplysia CPEB has prion-like properties. Cell, ۱۱۵، ۸۷۹–۸۹۱، (۲۰۰۳)
