نشانگان اهلرز-دنلوس
نشانگان اهلرز-دنلوس (به انگلیسی: Ehlers–Danlos syndrome) گروهی از اختلالات ارثی نادر و ناهمگون بافت همبند هستند که در آن نقائص ژنتیکی موجب بروز مشکلاتی در تولید، ساختمان یا پروسس کلاژن میشود. این اختلالات با افزایش محدودهٔ حرکات مفصل، شکنندگی و افزایش قابلیت ارتجاعی پوست مشخص میشود. تشخیص اغلب دشوار میباشد و بیشتر بر معیارهای بالینی و سابقهٔ فامیلی استوار است.
| نشانگان اهلرز-دنلوس | |
|---|---|
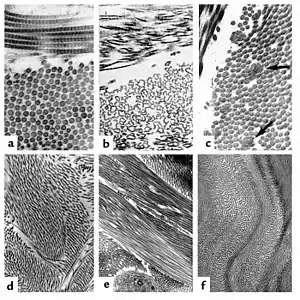 | |
| The collagen fibril and EDS. (a) Normal collagen fibrils are of uniform size and spacing. Fibrils from a patient with dermatosparaxis (b) show dramatic alterations in fibril morphology with severe effects on tensile strength of connective tissues. Patients with classical EDS (c) show composite fibrils. Fibrils from a TNX-deficient patient (d) are uniform in size and no composite fibrils are seen. TNX-null (e) fibrils are less densely packed and not as well aligned to neighboring fibrils. | |
| طبقهبندی و منابع بیرونی | |
| تخصص | ژنشناسی پزشکی |
| آیسیدی-۱۰ | Q79.6 ( ILDS Q82.817) |
| آیسیدی-9-CM | 756.83 |
| مدلاین پلاس | 001468 |
| ئیمدیسین | derm/۶۹۶ ped/654 |
| پیشنت پلاس | نشانگان اهلرز-دنلوس |
| سمپ | D004535 |
تاریخچه
این سندرم برگرفته از نام دو پزشک به نامهای " ادوارد اهلر " از دانمارک و "هنری الکساندر دانلوس" از فرانسه میباشد.


کلاژن
کلاژن پروتئینی است که در ماتریکس خارج سلولی جانوران، وجود دارد. کلاژن فراوانترین پروتئین بدن محسوب میگردد. دست کم ۱۳ نوع کلاژن در انسان شناسایی شدهاند که هر کدام در بافت خاصی وجود دارند. انواع کلاژن:
کلاژن نوع I: این نوع کلاژن در استخوان و وتر وجود دارد و از تعداد زیادی دستههای فیبری ساخته شدهاست که به صورت امواج دریا قرار دارند.
کلاژن نوع II: که در غضروف شفاف دیده میشود به شکل فیبریل است و فیبر تشکیل نمیشود.
کلاژن نوع III: همان رشته شبکهای است که در غشای پایه دیده میشود این نوع کلاژن از فیبرهای واحد ساخته شدهاست.
کلاژنهای نوع V,IV: به صورت فیبریل نبوده و احتمالاً غیر پلیمریزه هستند. کلاژن نوع IV در غشای پایه ونوع V در جدار رگهای خونی جنین یافت میشود.
بیماریزایی
علائم این سندرم بستگی به نوع کلاژن درگیر دارد مانند مفاصل سست و لغزنده، هیپرالاستیسیتی پوست، ضعف بافتی، تأخیر در ترمیم زخم، کبودی آسان و دررفتگی مکرر مفاصل. نوع هایپرموبیلیتی (درگیری کلاژن تیپ ۳)، نوع کلاسیک (تیپ ۱ و ۲)، عروقی (تیپ ۴)، کیفواسکلیوز (تیپ ۶) و آرتروکالازیا تیپ ۷ از انواع این سندرم هستند که مشکلات در گونههای مختلف کلاژن است مثلاً در نوع کلاسیک (تیپ ۱ و ۲) اختلال در کلاژن نوع ۵ است. اغلب مانند نوع کلاسیک اتوزومال غالبند و برخی مانند نوع کیفواسکلیوزیس اتوزومال مغلوب.
| نام | تیپ | توصیف | OMIM | ژن(ها) |
| بیش حرکتی | type 3 | Affects 1 in 10,000 to 15,000 and is caused by an autosomal dominant or autosomal recessive mechanism. Mutations in either of two separate genes (which are also involved in Vascular EDS and Tenascin-X deficiency EDS, respectively) may lead to this variant. Joint hypermobility is the hallmark of this type, with less severe skin manifestations. Joint instability and chronic musculoskeletal pain are particularly prominent in this type. Patients with the Hypermobility Type experience frequent joint dislocations and subluxations (partial/incomplete dislocations), with or without trauma. As a result, pain is a common, severe, and a lifelong symptom of this type. Additionally, osteoarthritis is common, and many get it earlier in life than expected.[1] | 130020 | COL3A1، TNXB |
| کلاسیک | types 1 & 2 | Affects approximately 1 in 20,000 to 50,000 people. It is caused by autosomal dominant mechanism and affects type-V collagen, as well as type I. Type 1 typically presents with severe skin involvement, and type 2 presents with mild to moderate skin involvement. Patients with the Classical Type may experience the same symptoms as the Hypermobility Type. The main difference between the Hypermobility and Classical Types are the Classical has more skin involvement while the Hypermobility Type has more joint involvement. Those with Classical EDS can also have severe joints issues like those with the Hypermobility Type. | 130000، 130010 | COL5A1، COL5A2، COL1A1 |
| عروقی | type 4 | Is an autosomal dominant defect in the type-III collagen synthesis; affecting approximately 1 in 100,000 to 250,000 people. The vascular type is considered one of the more serious forms of Ehlers–Danlos syndrome because blood vessels and organs are fragile and prone to tearing (rupture). Many patients with EDS type 4 express a characteristic facial appearance (large eyes, small chin, sunken cheeks, thin nose and lips, lobeless ears), have a small stature with a slim build, and typically have thin, pale, translucent skin (veins can usually be seen on the chest and abdomen) with very easy bruising and propensity to develop ecchymoses (bruising without trauma). About one in four people with vascular type EDS develop a significant health problem by age 20 and more than 80 percent develop life-threatening complications by age 40. | 130050 | COL3A1 |
| کیفواسکلیوزیس | type 6 | Is an autosomal recessive defect due to deficiency of an enzyme called lysyl hydroxylase; it is very rare, with fewer than 60 cases reported. The kyphoscoliosis type is characterised by progressive curvature of the spine (scoliosis), fragile eyes, and severe muscle weakness. | 225400، 229200 | PLOD1 |
| Arthrochalasiaآرتروکالازیا | types 7A & B | Is also very rare, with about 30 cases reported. It affects type-I collagen. The arthrochalasia type is characterised by very loose joints and dislocations involving both hips. Their joints are much looser than the Hypermobility Type. It could be considered 2+ times worse than someone who has severe joint instability with the Hypermobility Type. | 130060 | COL1A1، COL1A2 |
| Dermatosparaxis | type 7C | Also very rare, with about 10 cases reported. The dermatosparaxis type is characterised by extremely fragile and sagging skin. | 225410 | ADAMTS2 |
درمان
چون بروز بیماری به علت موتاسیون ژنی میباشد بنابراین درمان قطعی برای این سندرم وجود ندارد و درمان بیشتر به تسکین علائم و عوارض ناشی از این سندرم میپردازد.
جستارهای وابسته
منابع
- Levy, Howard (2004). “The Ehlers Danlos Syndrome, Hypermobility Type. ” University of Washington: NIH. Retrieved from http://newtons-online.net/documents/EDS%20b.pdf بایگانیشده در ۱۹ اکتبر ۲۰۱۳ توسط Wayback Machine
- مشارکتکنندگان ویکیپدیا. «syndrome=۳۶۳۲۲۴۱۳۵ Ehlers–Danlos syndrome». در دانشنامهٔ ویکیپدیای انگلیسی، بازبینیشده در ۲۰۱۴-۰۶-۰۱.