پروفایل بیان ژن
در حوزهٔ زیست مولکولی، اندازهگیری پروفایل بیان ژن معیاری از فعالیت هزاران ژن به صورت یکجا، برای ایجاد یک تصویر عمومی از کارکرد سلول است. این پروفایل میتواند برای مثال بین سلولهای فعال جدا از هم تمایز ایجاد کند، یا مشخص کند یک سلول چگونه به یک درمان واکنش نشان میدهد. بسیاری از آزمایشهای در این سطح، کل ژنوم را به صورت همزمان، برای هر ژن، در یک سلول خاص اندازهگیری میکنند.
تعداد زیادی تکنولوژی ترانسکریپتوم برای تولید دادهٔ مورد نیاز برای آنالیز میتواند مورد استفاده واقع شود. ریزآرایهٔ دیاِناِی[1] فعالیت نسبی یکسری ژن هدف را اندازه میگیرد. تکنولوژیهای مبتنی بر دنباله، مانند توالییابی آرانای علاوه بر سطح بیان ژنها، اطلاعاتی از دنبالهٔ آنها را نیز فراهم میکنند.
مقدمه
بدست آوردن بیان ژن، مرحلهٔ منطقی پس از توالییابی ژنوم است: دنبالهٔ ژنوم به ما این اطلاعات را میدهد که سلول چه فعالیتهایی انجام میدهد، در حالی که پروفایل بیان ژن مشخص میکند دقیقاً در آن لحظه چه کارهایی انجام میشود. ژنها شامل دستورهایی هستند که آراِناِیهای پیامرسان (mRNA) را میسازند، اما در هر لحظه هر سلول فقط بخشی از ژنهایی که دارد را به mRNA تبدیل میکند. چنانچه ژنی، در حال تولید mRNA باشد، آن ژن «روشن» و در غیر این صورت «خاموش» در نظر گرفته میشود. معیارهای زیادی مشخص میکنند که یک ژن روشن یا خاموش باشد، از جملهٔ آنها میتوان به زمان، محیطی که در آن قرار دارد و سیگنالهای شیمیایی که از سلولهای دیگر دریافت میکند اشاره کرد.
بررسی پروفایل بیان ژن، معمولاً اندازهٔ نسبی بیان mRNAها را در دو یا چند شرایط آزمایشگاهی بدست میآورد. به این دلیل که تغییرهای سطح بیان یک دنبالهٔ مشخص از mRNA، تغییرهایی در پروتئین حاصل از آن ژن را نشان میدهد، که میتوان نمایندهٔ یک شرایط آسیبدیده یا پاسخ همایستایی باشد. برای مثال سطح بیان بالای mRNA ای که الکل dehydrogenase را کد میکند نشاندهندهٔ این است که سلولها یا بافت مورد بررسی، در حال پاسخگویی به افزایش سطح اتانول در محیط است.[2][3][4]
مقایسه با پروتئومیک
ژنوم انسان شامل حدود ۵۰۰۰ ژن میباشد که در حدود ۱٬۰۰۰٬۰۰۰ پروتئین متفاوت را تولید میکنند. این بهخاطر پیرایش جایگزین است و همینطور به خاطر اینکه سلولها تغییرهایی را پس از اینکه بار اول پروتئین را ایجاد کردند، در پروسهٔ پیرایش پسارونویسی ایجاد میکنند، و در نتیجه، هر ژن میتواند پایهای برای تولید نسخههای متفاوت پروتئین باشد. با یک آزمایش mass spectrometry میتوان حدود ۲۰۰۰ پروتئین یا ۰٫۲٪ از کل پروتئینها را شناسایی کرد.[5] اطلاعاتی که از خود پروتئینها (پروتئومیک) بدست میآید از اطلاعاتی که از RNAهای پیامرسان استخراج میشود دقیقتر است.[6]
کاربرد در تولید و آزمون فرضیه
در بعضی مواقع، یک دانشمند فرضیهای در ذهن دارد و در مورد اتفاقهایی که ممکن است بیفتد ایدههایی دارد، و با انجام آزمایشهای پروفایل بیان ژن سعی دارد این فرضیه را رد کند. در حقیقت دانشمند پیشبینیای در مورد سطح بیان سلولی میکند که ممکن است اشتباه باشد.
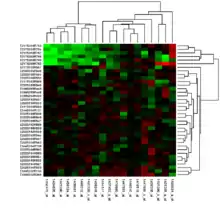
معمولاً پروفایل بیان ژن زمانی بدست آورده میشود که اطلاعات کاملی در مورد تعامل ژنها با شرایط آزمایشگاهی در مورد یک فرضیهٔ در حین آزمون نداریم. بدون فرضیه، چیزی برای انکار کردن یا اثبات کردن وجود ندارد، اما پروفایل بیان میتواند برای شناسایی فرضهای کاندید برای آزمایشهای آینده کمککننده باشد. بسیاری از آزمایشهای پروفایل بیان ژن ابتدایی و امروزی، به صورت «اکتشاف کلاس» شناسایی میشوند که به فرمی است که در ادامه توضیح داده میشود.[7] روش معروف برای اکتشاف کلاس شامل گروهبندی کردن ژنها یا نمونههای مشابه با استفاده از خوشهبندی کی_میانگین یا سلسلهمراتبی میباشد. مستقل از روش خوشهبندیای که مورد استفاده قرار میگیرد، نیاز است کاربر معیار فاصلهٔ مناسبی (فاصله یا شباهت) بین دادهها برگزیند.[8] تصویر بالا خروجی خوشهٔ دوبعدی را نشان میدهد، به طوری که نمونههای مشابه (سطرها) و پروب ژنهای مشابه (ستون) به گونهای مرتب شدهاند که نزدیک هم قرار دارند. سادهترین شکل اکتشاف کلاس، لیست کردن همهٔ ژنهایی است که بین دو شرایط آزمایشگاهی بیشتر از یک حدی تغییر کردهاند.
پیشبینی کلاس از اکتشاف کلاس بسیار سختتر است، اما این امکان را به افراد میدهد که به سوالهای مهمی که مستقیماً بالینی هستند پاسخ دهد. برای نمونه با دادن این پروفایل امکان این که بیمار به یک داروی خاص پاسخ دهد چقدر است؟ پاسخ به این سؤال نیاز به تعداد زیادی نمونه از پروفایل دارد که به دارو پاسخ دادهاند و تعدادی که پاسخ ندادهاند.
اعتبارسنجی اندازهگیریهای با حجم بالا
هر دو تکنولوژی ریزآرایهٔ دیانای و واکنش زنجیرهای پلیمراز بلادرنگ (qPCR)، از جفتبازهایِ دنبالهٔ مکمل نوکلئیک اسید استفاده میکنند، و هر دو در بدست آوردن پروفایل بیان ژن، معمولاً در یک روش سریالی استفاده میشوند. در حالی که تکنولوژی حجم بالای ریزآرایهٔ دیاِناِی دقت qPCR را ندارد، اندازهگیری بیان ژن چندین ژن با استفاده از qPCR و اندازهگیری کل ژنوم با استفاده از ریزآرایهٔ دیانای تقریباً زمان یکسانی میبرد؛ بنابراین منطقی است که ابتدا آزمایشهای تحلیل ریزآرایهٔ دیانایِ شبه کمّیای برای شناسایی ژنهای کاندید انجام شود، سپس qPCR روی تعدادی از ژنهای جالب بدست آمده در مرحلهٔ قبل انجام شود.
تفسیر ژن
در حالی که ممکن است آمار شناسایی کند کدام محصول از ژن تحت شرایط آزمایشگاهی تغییر میکند، تفسیر و درک ابعاد زیستی بیان ژن بستگی به این دارد که بدانیم هر پروتئین از کدام ژن بدست آمدهاست و چه عملکردهایی دارد. تفسیر ژن اطلاعات عملکردی و دیگر اطلاعاتی مانند مکان ژن در کروموزوم را فراهم میکند. بعضی از تفاسیر عملکردی نسبت به سایر تفاسیر قابل اعتمادتر هستند. پایگاه دادهٔ تفسیر ژن به صورت مرتبط تغییر میکند و پایگاه دادههای متفاوت به یک پروتئین یکسان با نامهای متفاوتی اشاره میکنند، و نشاندهندهٔ این است که درک عملکرد پروتئین مدام در حال تغییر است.[9][10]
دستهبندی ژنهای تنظیمشده
پس از شناسایی مجموعهٔ ژنهای تنظیمشده، مرحلهٔ بعدی بدست آوردن پروفایل بیان ژن است که در برگیرندهٔ جستجوی الگویی بین مجموعهٔ تنظیم شدهاست. سؤالی که مطرح میشود این است که آیا پروتئینهایی که از این ژنها بدست میاد عملکردهای مشابهی دارند؟ آیا از نظر شیمیایی مشابه هستند؟ آیا در موقعیت مشابهی از سلول قرار دارند؟ تحلیل هستیشناسی ژن روش استانداردی برای تعریف این روابط میباشد. هستیشناسی ژنها با دستهبندی بسیار وسیعی شروع میشود، برای مثال «فرایند سوختوساز» و آنها را به دستههای کوچکتر تقسیم میکند.
ژنها در کنار عملکرد زیستی، ویژگیهای شیمیایی، و موقعیت سلولی، ویژگیهای دیگری هم دارند. میتوان مجموعهای از ژنها را برحسب میزان نزدیکیشان به ژنهای دیگر با هم ترکیب کرد و ارتباطشان با یک بیماری، داروها یا سمها را بدست آورد.
یافتن الگویی بین ژنهای تنظیمشده
ژنهای تنظیمی با توجه به اینکه چه کاری انجام میدهند و چه هستند دستهبندی میشوند، و ممکن است ارتباطهای مهمی بین ژنها بدست آورده شود. برای نمونه، ممکن است شواهدی صورت گیرد که یک ژن خاص، پروتئینی میسازد که آن یک آنزیم را ایجاد میکند و آنزیم تولید شده، پروتئین دیگری را فعال میکند که باعث میشود ژن دوم دیگری در لیست روشن شود. این ژن دوم ذکر شده ممکن است یک سازه رونویسی باشد که ژن دیگری از لیست ما را تنظیم میکند. مشاهدهٔ این ارتباطها، ما را مشکوک میکند که این لیست، ارتباطهای فراتر از شانس با یکدیگر دارند و همهٔ آنها به خاطر یک فرایند زیستی پایه در لیست ما قرار گرفتهاند. از طرفی اگر چند ژن به صورت رندم انتخاب کنیم، میتوان تعدادی ژن مرتبط با آنها پیدا کرد. در این موارد، به پردازشهای آماری دقیقی نیاز داریم تا مشخص شود آیا نتیجهٔ زیستی بدست آمده معنادار هست یا نه. در این شرایط تحلیل مجموعهٔ ژنها مورد نیاز است.
رابطهٔ علل و معلول
ابتدائاً تحلیلهای آماری ساده مشخص میکند آیا ارتباط بین ژنهای درون لیست بیشتر از مقداری است که میتواند شانسی باشد. این آمار حتی در صورتی که بیش از حد سادهسازی شده باشد، میتواند جالب باشد. یک مثال را بررسی میکنیم، فرض کنیم در یک آزمایش ۱۰۰۰۰ ژن داریم، فقط ۵۰ درصد آنها نقشی در ساختن کلسترول بازی میکنند. آزمایش ۲۰۰ ژن تنظیمی را تشخیص میدهد. در بین این ۲۰۰ ژن ۴۰ عدد در بین لیست ژنهای تأثیرگذار در کلسترول هستند. با توجه به تعداد ژنهای کلسترول در کل (۰٫۵٪)، انتظار میرود به ازای هر ۲۰۰ ژن، یکی از آنها جزو ژنهای تأثیرگذار در کلسترول باشد، که برابر است با ۰٫۰۰۵ برابر ۲۰۰. این پیشبینیِ مورد انتظار است و ممکن است کسی بیشتر از یک ژن مشاهده کند. سؤال این است که چه زمانی ما میتوانیم به صورت شانسی به جای ۱ ژن ۴۰ ژن مشاهده کنیم.
با توجه به توزیع فوق هندسی، انتظار میرود باید حدود ۵۷^۱۰ بار لیست ۲۰۰ تایی ژن به صورت تصادفی انتخاب شود تا بتوان یک لیست شامل ۳۹ یا بیشتر، ژن مشترک با لیست تأثیرگذار در کلسترول در آن پیدا کرد. چنانچه ممکن است از جنبهای، مشاهدهٔ تصادفی این مورد بسیار کوچک به نظر آید، ممکن است کسی نتیجه بگیرد که لیست ژنهای تنظیمی، بسیار غنی از ژنهای مربوط به کلسترول هستند.[11]
ممکن است فرض شود اِعمال شرایط خاص در آزمایش، باعث تنظیم کلسترول شدهاست، زیرا نحوهٔ درمان به نظر گونهایست که ژنهای مربوط به کلسترول را تنظیم میکند. در حالی که ممکن است این فرضیه درست به نظر برسد، دلایلی وجود دارد که نتیجهگیری بر اساس غنی بودن به تنهایی، نتیجهٔ غیرقابلتوجیهی را میدهد.
نتیجهگیری
بدستآوردن پروفایل بیان ژن، اطلاعات جدیدی در مورد اینکه ژنها در شرایط متفاوت چه کارهایی میکنند به ما میدهد.[12] بهطور کلی، تکنولوژی ریزآرایه، پروفایل بیان ژن قابل اطمینانی فراهم میکند. با استفاده از این اطلاعات میتوان فرضیههای جدیدی در مورد واقعیتهای زیستی یا آزمایشی بدست آورد. اگرچه اندازه و پیچیدگی این آزمایشها معمولاً منجر به انواع متنوعی از تفاسیر میتواند بشود. در بسیاری از موارد، نتیجهٔ تحلیل پروفایل بیان ژن بسیار زیاد بیشتر از خود آزمایش اولیه زمان میبرد.
بسیاری از محققین روشهای مختلف آماری و تحلیل دادهٔ اکتشافی را قبل از انتشار نتایج پروفایل بیان ژن، استفاده میکنند، و تلاشهایشان را با بایوانفورماتیستها یا دیگر متخصصان در ریزآرایهٔ دیانای هماهنگ میکنند. یک طراحی آزمایش خوب، تکرار کافی زیستی و پیگیری آزمایش، نقشهای مهمی در موفقیت آزمایشهای پروفایل بیان ژن دارند.
منابع
- "Microarrays Factsheet". Retrieved 2007-12-28.
- Suter L, Babiss LE, Wheeldon EB (2004). "Toxicogenomics in predictive toxicology in drug development". Chem. Biol. 11 (2): 161–71. PMID 15123278. doi:10.1016/j.chembiol.2004.02.003.
- Magic Z, Radulovic S, Brankovic-Magic M (2007). "cDNA microarrays: identification of gene signatures and their application in clinical practice". J BUON. 12 Suppl 1: S39–44. PMID 17935276.
- Cheung AN (2007). "Molecular targets in gynaecological cancers". Pathology. 39 (1): 26–45. PMID 17365821. doi:10.1080/00313020601153273.
- Mirza SP, Olivier M (2007). "Methods and approaches for the comprehensive characterization and quantification of cellular proteomes using mass spectrometry". Physiol Genomics. 33 (1): 3–11. PMC 2771641 Freely accessible. PMID 18162499. doi:10.1152/physiolgenomics.00292.2007.
- Hebert AS, Richards AL, et al. (2014). "The One Hour Yeast Proteome". Mol Cell Proteomics. 13 (1): 339–347. doi:10.1074/mcp.M113.034769.
- Chen JJ (2007). "Key aspects of analyzing microarray gene-expression data". Pharmacogenomics. 8 (5): 473–82. PMID 17465711. doi:10.2217/14622416.8.5.473.
- Jaskowiak, Pablo A; Campello, Ricardo JGB; Costa, Ivan G (24 January 2014). "On the selection of appropriate distances for gene expression data clustering". BMC Bioinformatics. 15 (Suppl 2): S2. PMC 4072854 Freely accessible. PMID 24564555. doi:10.1186/1471-2105-15-S2-S2.
- Dai M, Wang P, Boyd AD, et al. (2005). "Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data". Nucleic Acids Res. 33 (20): e175. PMC 1283542 Freely accessible. PMID 16284200. doi:10.1093/nar/gni179.
- Alberts R, Terpstra P, Hardonk M, et al. (2007). "A verification protocol for the probe sequences of Affymetrix genome arrays reveals high probe accuracy for studies in mouse, human and rat". BMC Bioinformatics. 8: 132. PMC 1865557 Freely accessible. PMID 17448222. doi:10.1186/1471-2105-8-132.
- Curtis RK, Oresic M, Vidal-Puig A (2005). "Pathways to the analysis of microarray data". Trends Biotechnol. 23 (8): 429–35. PMID 15950303. doi:10.1016/j.tibtech.2005.05.011.
- Couzin J (2006). "Genomics. Microarray data reproduced, but some concerns remain". Science. 313 (5793): 1559. PMID 16973852. doi:10.1126/science.313.5793.1559a.
