نشانگان سرطان
نشانگان سرطانی یا نشانگان سرطان خانوادگی، یک آشفتگی و اختلال ژنتیکی است در آن کسی که جهشهای ژنی در یک یا بیشتر ژنهای تکی مستعدی که تغییر یافتهاند برای پیشروی و پیشرفت سرطان را به ارث برده و میتواند منجر به تاخت و تاز و هجوم دیگر سرطانها نیز شود. نشانگانهای سرطان معمولاً بهتنهایی یک خطر زندگی بزرگ پیشروی سرطان ظاهر نمیشوند، ولی پیشرفت چندگانه وابسته به تومورهای نخستین دانسته میشوند.[1] بسیاری از این نشانگانها بهواسطهٔ جهشهایی در ژنهای سرکوبگر تومور رخ میدهد که این ژنها در فراهم آوری سلولهای توموری سرطاندار نقش دارند. دیگر ژنهایی که میتوانند در تعمیر دیانای مؤثر باشند؛ آنکوژنها و ژنهایی هستند که در ساخت رگهای خونی (رگزایی) شرکت میکنند.[2] نمونههای رایج از نشانگانهای سرطان ارثی، نشانگان سرطان پستان – تخمدان و سرطان روده بزرگ بی پولیپوزیز ارثی (نشانگان لینچ[برابرها 1]) میباشند.[3][4]
پیشزمینه
نشانگانهای سرطان ارثی زیر ۵٪ تا ۱۰٪ همهٔ سرطانها قرار دارند.[5] دانستن علمی یک سرطان؛ آمادگی نشانگانهای که بهطور فعال بسط یافتهاند: نشانگانهای زاید و اضافی یافت شدند،[5] زیستشناسی، اساساً واضحتر شد و تجاریسازی روششناسی ژنتیک تشخیصی در بهبودی بیماریهای بالینی انجام یافت. شیوع سرطانّهای پستان و رودهٔ بزرگ که بیشترین نشانگانهای بازشناختی را داراست و شامل نشانگان سرطان تخمدان – پستان ارثی[برابرها 2] و سرطان روده بزرگ نا-پولیپوزیز ارثی[برابرها 3] (نشانگان لینچ) میباشد.[5]
برخی سرطانهای کمیاب و نادر با نشانگانهای آمادگیکنندهٔ سرطان ارثی قویا درآمیختهاند. آزمونهای ژنی شاید بتوانند با کارسینومای فوق کلیوی[برابرها 4] با تومورهای سرطانواره[برابرها 5]؛ سرطان لیومیوسارکوم[برابرها 6]؛ سرطان مغزینهای تیروئید[برابرها 7]؛ سرطان پاراگانگلیون[برابرها 8] یا فئوکروموسیتوما[برابرها 9]؛ کارسینومای سلول کلیوی رنگگریز[برابرها 10]؛ آنکوسیتیک دورگهی[برابرها 11] یا بافتشناسی سرطان آنکوسیتی[برابرها 12]؛ کارسینومای چربی[برابرها 13]؛ و تومورهای طناب جنسی با لولههای حلقوی[برابرها 14].[5][6]
دانش ژنتیک
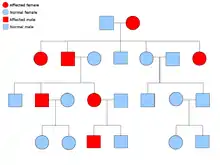
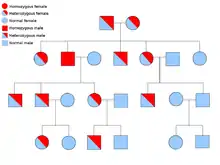
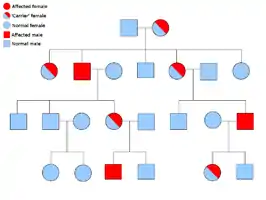
دو نسخهٔ از هر ژنی که در تمام یاختههای بدن هر کسی حضور دارد را «آلل»[برابرها 15] مینامند. بیشتر نشانگانهای سرطان در یک راه مندلی اتوزومی غالب پخش میشود. در این موارد، تنها یک آلل معیوب میتوان بهتنهایی سرطان را در شخص ایجاد نماید. کسانی که دارای چنین آللی هستند را «هتروزیگوس» و کسانی که دارای دو آلل یکسان چه معیوب-معیوب و چه سالم-سالم را «هموزیگوس» خوانند. برپایه ژنتیک مندلی از آمیزش یک هتروزیگوس با یک هموزیگوس، زادههای حاصل ۵۰٪ احتمالاً دارای آن بیماری هستند.[6] جهش در ژن به ارث برده شده بانام «یک جهش جنینی»[برابرها 16] شناخته میشود و یک جهش بیشتر در یک آلل طبیعی در پیشرفت و توسعهٔ سرطان نتیجه میدهد. این رخداد بانام «فرضیهٔ دو ضربهای کنودسون»[برابرها 17] شناخته میشود که نخستین ضربه به ژن یک ژن ارثی است و دومین در زندگی رخ میدهد.[2] همانگونه که یک آلل نیازمند این است که جهش یافته شود (قیاسا هر دوی آنها را «سرطانهای پراکنده»[برابرها 18] میخوانند)، احتمال جهش تکی بیشتر از احتمال دو جهش پیاپی در جمعیت است.
کمترین احتمال، نشانگانهایی هستند که به صورت آتوزومی بازگشتی در دودمانه پخش میشوند. جفت آللهای ژن بایستی جهشیافته باشد در اختلالات بازگشتی آتوزومی برای آمادهسازی آن به سرطان است. کسی که با دو آلل بازگشتی است به عنوان یک «هموزیگوس بازگشتی»[برابرها 19] دانسته میشود. هر دوی والدین بایستی یک آلل معیوب و یک طبیعی داشته باشند تا زادهای هموزیگوس بازگشتی پدیدآورند. اگر والدین دارای یک آلل جهشیافته و یک آلل طبیعی (هتروزیگوس) باشند، ۲۵٪ احتمال زادهٔ هوموزیگوس بازگشتی، ۲۵٪ احتمال زادهٔ هتروزیگوس که حامل ژن معیوب است و ۲۵٪ احتمال زادهٔ هوموزیگوس سالم و طبیعی را دارند.[6]
نمونههای نشانگانهای سرطان آتوزومی غالب: نشانگان خودایمنی تکثیر لمفوسیتی[برابرها 20] که بانام نشانگان کانل – اسمیت[برابرها 21] شناخته میشود؛ نشانگان بکواید – ویدمان[برابرها 22] که ۸۵٪ موارد سرطانهای پراکنده هستند؛ نشانگان بیرت – هوگ – دوبه[برابرها 23]؛ نشانگان کانری[برابرها 24]؛ نشانگان کودِن[برابرها 25]؛ سرطان نخاع خانوادگی[برابرها 26]؛ نشانگان خال دیسپلازی با سرطان ملانین،[برابرها 27] پولیپوزیس غدهای خانوادگی[برابرها 28]؛ نشانگان سرطان پستان – تخمدان ارثی؛ سرطان معدی پخششوندهٔ ارثی[7]؛ سرطان رودهبزرگ نا پولیپوزیس ارثی یا نشانگان لینچ؛ نشانگان لی – فرومنی[برابرها 29]؛ نئوپلازیای درونریز چندگانهٔ تیپ ۲/۱[برابرها 30]؛ استئوکندروماتوزیز چندگانه[برابرها 31]؛ نشانگان پوتز – جگرز[برابرها 32]؛ سرطان پروستات خانوادگی[برابرها 33]؛ سرطان یاختهٔ کلیوی لیومیوماتوزیز ارثی[برابرها 34]؛ سرطان سلول کلیوی برآمدگی ارثی[برابرها 35]؛ نشانگان پاراگانگلیوما – فنوکروموسیتوما ارثی[برابرها 36]؛ تومور شبکیه[برابرها 37]؛ تصلب تکمهای[برابرها 38] بیماری فون هیپل – لینداو[برابرها 39] و تومور ویلم.[برابرها 40][8]
نمونههای نشانگانهای سرطانی بازگشتی آتوزومی: آتاکسی تلانژیکتازی،[برابرها 41] نشانگان بلوم،[برابرها 42] کمخونی فانکُنی،[برابرها 43] پلیپوزیس همبسته با MUTYH، نشانگان روتموند – تامسون،[برابرها 44] نشانگان ورنر[برابرها 45] و خشکپوستی رنگدانهای[برابرها 46].[8]
برخی نمونهها
گرچه نشانگانهای سرطانی یک خطر افزایش یافته سرطان را نمایش میدهند ولیکن خطر متفاوت است. برای برخی از آن بیماریها، سرطان در آغاز بروز نمیکند. در اینجا بحث بر روی خطر افزایشی سرطان همراه آن متمرکز شدهاست. این سیاهه به دور از پرچانگی و توضیح زیاد است.
کمخونی فانکنی
کمخونی فانکنی (FA) یک اختلال همراه با طیفی پهناور نشانههای بالینی است که شامل: آغاز زودرس و افزایش خطر سرطان، نقصان مغز استخوان و ناهنجاریهای مادرزادی میباشد. برجستهترین ظهور این اختلال وابستگیاش به خونسازی تولید خون به واسطهٔ مغز استخوان) بوده که دربردارندهٔ: کمخونی آپلاستیک،[برابرها 47] نشانگان میلودیزپلاستیک[برابرها 48] و لِکومیا میلوئید حاد[برابرها 49] است. تومورهای کبدی و سرطان سلولهای سنگفرشی[برابرها 50] مری، گلو و زبان کوچک معمولاً تومورهای سخت و سفت در ارتباط با کمخونی فانکنی هستند. ناهنجاریهای مادرزادی شامل: ناهنجاریهای استخوانی (به ویژه بر روی دستها اثر میکند)، خالهای کافه آو لایت[برابرها 51] و هیپوپیگمانتاسیون[برابرها 52] هستند. امروزه، ژنهای شناخته شدهای که منجر به کمخونی فانکنی میشوند، عبارتاند از: FANCA, FANCB, FANCC, FANCD2، FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN, FANCO, FANCP و BRCA2 (پیشتر با نام FANCD1 شناخته میشد). ارث این نشانگان عمدتاً آتوزومی بازگشتی است ولی FANCB میتواند از کروموزومهای x پدری یا مادری (ارثیت بازگشتی x-پیوندی) به ارث برسد. مسیر کمخونی فانکنی در تعمیر DNA درگیر میشود، هنگامی که دو رشتهٔ DNA به صورت نادرست بهم الحاق میشوند (کراس لینکهای درونرشتهای). بسیاری مسیرها با مسیر کمخونی فانکنی هماهنگ میشوند از اینرو شامل تعمیر بُرِش نوکلئوتید، سنتز ترجمه و نوترکیبی همتا میباشند،[9]،[10]،[11]،[12]،[13][14]
پولیپوزیس غدهای خانوادگی
پولیپوزیس غدهای خانوادگی (FAP) یک نشانگان غالب آتوزومی است که خطر سرطان روده-مقعدی را بهطور زیادی افزایش میدهد. از هر ۸۰۰۰ تن، یکی این بیماری را داشته و این بیماری تقریباً نافذیت ۱۰۰٪ دارد. فرد دچار بیماری در رودهٔ بزرگ خود صدها هزار از این غدههای سرطانی را خواهد داشت و در بیشتر موارد به سرطان مترقی و پیشرونده تغییر میکند. دیگر تومورهای افزایش یافته در فراوانی شامل: سرطانهای استخوانی،[برابرها 53] سرطانها و کارسینوماهای غدههای فوق کلیوی، تومورهای تیروئید و تومورهای دسموئید[برابرها 54] هستند. عامل این اختلال یک ژن APC جهشیافتهاست که در تنظیمβ-catenin شرکت میکند. APC معیوب منجر به انباشتن β-catenin در یاختهها و فعالسازی عوامل رونویسی درگیر در تکثیر، مهاجرت، تمایز و مرگ برنامهریزی شده سلولی یا آپوپتوز میشود،[15]،[16].[17]
سرطان تخمدان و پستان ارثی
نشانگان سرطان پستان – تخمدان ارثی (HBOC) یک اختلال ژنی آتوزومی غالب که با جهشهای ژنی بر ژنهای BRCA1 و BRCA2 رخ میدهد. در زنان این اختلال عمدتاً خطر سرطان پستان و تخمدان را افزایش میدهد ولی خطر کارسینومای لولهٔ فالوپ و کارسینومای خونابهدار برآمدهٔ صفاق[برابرها 55] را نیز افزایش میدهد. در مردان خطر سرطان پروستات افزایش مییابد. دیگر سرطانهایی که متناقضانه با این نشانگان پیوند خوردهاند؛ سرطان لوزالمعده، سرطان پستان نرینه، سرطان روده بزرگ – مقعدی و سرطانهای گردن رحم و رحم. جهشهای ژنی تقریباً ۷٪ و ۱۴٪ سرطان پستان و تخمدان، به ترتیب، BRCA1 و BRCA2، ۸۰٪ موارد را تخمین میزنند. BRCA1 و BRCA2 هر جفت ژنهای سرکوبگر تومور مستلزم شده در بقا و تعمیر DNA هستند. جهشها بر روی این ژنها اجازه کمک به تخریب DNA را داده که میتواند منجر به سرطان شود،[18][19]
سرطان رودهٔ بزرگ نا-پولیپوزیس ارثی
سرطان رودهٔ بزرگ نا-پولیپوزیس ارثی (HNPCC) که بانام نشانگان لینچ نیز شناخته میشود، یک نشانگان سرطان غالب آتوزومی است که خطر سرطان روده بزرگ – مقعدی را افزایش میدهد. این سرطان با جهشهایی ژنی در تعمیر ناجور DNAیا MMR رخ میدهد، ژنهای شایان توجه MLH1، MSH2، MSH6 و PMS2 هستند. افزون بر سرطان رودهٔ بزرگ – مقعدی، فراوانی بسیاری سرطانهای دیگر نیز افزایش مییابد. آنها عبارت اند از: سرطان اندوتلیال، سرطان معده، سرطان تخمدان، سرطانهای رودهٔ کوچک و سرطان لوزالمعده. همچنین HNPCC با یک هجوم آغازین سرطان رودهٔ بزرگ – مقعدی همراه شدهاست. ژنهای MMR در تعمیر DNA هنگامی که بازها بر روی هر رشته از DNA جور نشود، درگیر میشوند. ژنهای MMR معیوب اجازه الحاق ممتد و جهشهای حذف در مناطقی از DNA که با عنوان ریزماهوارهها[برابرها 56] شناخته میشود را میدهد. این توالیهای کوتاه تکراری DNA ناپایدار گشته، منجر به یک حالت نااستواری ریزماهواره[برابرها 57] میشود. ریزماهوارههای جهشیافته معمولاً در ژنهای درگیر در راهاندازی و پیشرفت تومور یافت میشوند و MSI میتواند ابقای زیستی یاختهها را بالا برده و منجر به سرطان گردد،[4][20]،[21][22]
نشانگان پاراگانگلیونوما – فنوکروموسیتومای ارثی
بیشتر موارد پاراگانگلیونومای خانوادگی با جهشهایی در ژنهای (SDHD, SDHAF2, SDHC, SDHB) زیرواحد آنزیم سوکسینات دهیدروژناز یا SDH (succinate :ubiquinone oxidoreductase) رخ میدهد. جهش SDHDهمراه با PGL-1 همبسته شده و اکثراً PGL-1 به تنهایی با پاراگانگلیونومای پدری بیشتر از مادری مؤثرتر است. PGL1 و PGL2 با هم دیگر غالب همبارز هستند. PGL-4 با جهش SDHB همبسته شد و خطر بیشتر فنوکروموسیتوما و به همین روال سرطان سلول کلیوی و سرطان تیروئید نا-مغزینهای (non-medullary thyroid cancer) را بالا میبرد.[23]
نشانگان لی – فرومنی
نشانگان لی – فرومنی یک سندرم غالب آتوزومی است که معمولاً با جهشهایی در ژن TP53 رخ میدهد، که منجر به افزایش زیاد خطر سرطانهای بسیاری و نیز شروع زودتر همراه با سرطان میشود. سرطانهای در پیوند با این اختلال شامل: سارکوماهای بافتی نرم (در کودکی بیشتر یافت میشود)، اوسئوسارکوما، سرطان پستان، سرطان مغز، لِکِمیا (leukaemia) و کارسینومای قشر فوق کلیوی (adrenocortical carcinoma). افراد با نشانگان لی – فرومنی معمولاً دارای سرطانهای ابتدایی ناوابستهٔ چندگانه هستند. دلیل برای این طیف بزرگ بالینی از اختلالات شاید به خاطر جهشهای ژنی باشد که بیماری را تعدیل میکند. فراوردهٔ پروتئینی که با ژن TP53 ساخته شده، p53، در بازدارندگی چرخهٔ یاخته، تعمیر DNA و آپوپتوز درگیر است. p53 معیوب میتواند برای ایفای خالصانهٔ پردازشها توانا نباشد که شاید علتی برای تشکیل تومور باشد. زیرا به تنهایی ۶۰–۸۰٪ افراد دچار اختلال دارای جهشهای یافتنی در TP53 است، دیگر جهشها در مسیر p53 شاید در نشانگان لی – فرومنی شرکت کنند.[24][25][26][27]
پولیپوزیس همبسته شده با MUTYH
پولیپوزیس همبسته شده با MUTYH بیشترین جزئیات بالینی را با FAP به اشتراک میگذارد که تمایزی با یک اختلال آتوزومی بازگشتی را با جهشهایی درMUTYH ژن تعمیر DNA منجر میشود. تومورهای افزایش خطری در این اختلال، سرطان رودهٔ بزرگ – مقعدی، آدنوماهای معدی و آدنوماهای دوازدههای هستند.[28][29]
نشانگان کارسینومای سلول پایهای خالدار
نشانگان کارسینومای سلول پایهای خالدار (NBCCS) که همچنین با نام نشانگان گورلین (Gorlin syndrome) شناخته میشود، یک نشانگان سرطان غالب آتوزومی است که در هنگامی که کارسینومای سلول پایهای خطرش بسیار بالا باشد، رخ میدهد. بیماری با سلول خالدار پایهای، کراتوسیتها و ناهنجاریهای اسکلتی تشخیص داده میشود. تخمینهای شیوع NBCCS متفاوت بوده ولی تقریباً ۱ در ۶۰۰۰۰ است. حضور کارسینومای سلول پایهای در افراد سفیدپوست نسبت به سیاهپوست بسیار بالاتر بوده؛ به ترتیب ۸۰٪ و ۳۸٪ است. کراتوسیتهای دندانزایی (Odontogenic keratocysts) تقریباً در ۷۵٪ افراد دچار این بیماری یافت میشود و معمولاً در آغاز زندگی رخ میدهد. بیشترین ناهنجاریهای اسکلتی رایج در سر و چهره اتفاق میافتند ولی در دیگر مناطق گاهی مانند قفسه سینه نیز کارگر میشود. جهش ژنی مؤثرهٔ بیماری در ژن PTCH رخ میدهد و فراوردهٔ PTCH یک سرکوبگر تومور بوده که در پیامدهی سلول شرکت میکند. گرچه نقش صحیح این پروتئین در NBCCS ناشناخته است، ولی آن در مسیر پیامدهی جوجه تیغی (hedgehog signaling pathway) که در کنترل رشد و توسعه سلول شناخته شده، درگیر است.[30][31]
بیماری فون هیپل – لینداو
بیماری فون هیپل – لینداو (VHL) یک حالت نادر، ژنی غالب آتوزومی است که افراد دچار را مستعد به تومورهای بدخیم و خوشخیم میکند. بیشتر تومورهای رایج در VHL دستگاه عصبی مرکزی و همانژیوبلاستوماهای شبکیهای (retinal hemangioblastomas)، کارسینوماهای کلیوی سلولی تمیز (clear cell renal carcinomas)، فنوکروموسیتوماها، توموردارهای عصب-درونریز لوزالمعدی (pancreatic neuroendocrine tumours)، کیستهای لوزالمعدی، تومورهای کیسهٔ درون لمفاوی (endolymphatic sac tumors) و کیستادنوماهای نوکدار اپیدیدیمی. (epididymal papillary cystadenomas)[32][33] VHL از یک جهش در ژن سرکوبگر تومور فون هیپل – لینداو بر کروموزوم 3p25.۳ ناشی میشود.[34]
خشکپوستی رنگدانهدار
خشکپوستی رنگدانهدار (XP) یک اختلال بازگشتی آتوزومی است که با حساسیت بر پرتوی فرابنفش تشخیص داده میشود، خطر بزرگ افزایش آفتاب سوختگی و خطر افزایش سرطانهای پوست را منجر میشود. خطر سرطان پوست بیشتر از ۱۰۰۰ بار نسبت به افراد طبیعی است و شامل تیپهای سرطان پوست بسیاری همچون سرطانهای پوست ملانوما (melanoma) و نا-ملانوما (non-melanoma) است. همچنین، مناطق در معرض آفتاب، زبان، لبها و چشمها خطری افزایش یافتهای از سرطانی شدن را دارند. XP شاید با دیگر سرطانهای درونی و تومورهای خوشخیم همراه باشد. افزون بر سرطان، برخی جهشهای ژنی که منجر به XP میشوند با عصب-تباهی (neurodegeneration) همراه هستند. XP میتواند با جهشهای ژنی در ۸ ژن که پیرو آن فراوردههایی آنزیمی دارند، عبارت اند از: XPA, XPB, XPC, XPD, XPE, XPF, XPG و Pol η. XPA-XPF، آنزیمهای تعمیر برشی نوکلئوتید هستند که DNA آسیب دیده از پرتوی فرابنفش را تعمیر میکند و اجازهٔ ساخت جهشهای پروتئینهای معیوب را به واسطهٔ پرتوی فرابنفش میدهد. Pol η یک پلیمراز بوده که یک آنزیم درگیر در همانندسازی DNA است. پلیمرازهای بسیاری هستند ولیpol η آنزیمی است که DNA تخریب شده با پرتوی فرابنفش را همانندسازی میکند. افراد با جهشهای این ژن دارای یک زیرمجموعهٔ XP، واریانتی از بیماری XP هستند.[35][36]
تعمیر DNA و کاهش و افزایش خطر سرطان
بسیاری نشانگانهای سرطان به خاطر یک نقصان ارثی در توانایی تعمیر DNA است.[37] هنگامی که یک جهش ارثی موجود در یک ژن تعمیر DNA باشد، که ژن تعمیر در یک شکل دگرشیافته بیان خواهند شد یا نخواهند شد. سپس کنش تعمیر شبیه کاهش خواهد بود و نتیجتاً DNA آسیب میبیند و گرایش به انباشتگی بر خود خواهد داشت. همانگونه که آسیبهای DNA میتواند خطاهایی را باعث شود، سنتز DNA نیز منجر به جهشهایی میگردد؛ که برخی از آنها میتواند برانگیزندهٔ سرطان باشند. جهشهای تعمیر DNA خطی-اصلی (Germ-line repair mutations) که خطر سرطان را افزایش میدهند در جدول زیر فهرست بندی شدهاند:
| ژن تعمیر DNA | پروتئین | مسیرهای تعمیری مؤثر | سرطانها با خطر افزایش یافته |
| گشادشدگی مویرگهای آتاکسی[برابرها 58] جهشیافته | ATM | جهشهای گوناگون در ATM کاهشHRR, SSA یاNHEJ [38] | لوکمیا، لمفوما، سینه[39][40] |
| نشانگان شکوفه[برابرها 59] | BLM (هلیکاز) | HRR[41] | لوکمیا، لمفوما، روده بزرگ، سینه، پوست، شش، مجرای شنوایی، زبان، سرخنای، معده، لوزه، خشکنای، زهدان [42] |
| سرطان سینه ۱ و ۲ | BRCA1 BRCA2 | HRR دو رشتهای میشکند و رشتهٔ دختری شکافدار میگردد [43] | سینه، تخمدان[44] |
| ژنهای کمخونی فانکونی
FANCA,B،C,D1,D2,E,F,G,I,J,L,M,N,O,P |
FANCA etc. | HRR و TLS[45] | لوکمیا، تومورهای کبد، تومورهای جامد بسیاری مناطق[46] |
| ژنهای سرطان رودهای-مقعدی غیرپولیپوزیس ارثی MSH2 MSH6 MLH1 PMS2 | MSH2 MSH6 MLH1 PMS2 | MMR[47] | مشکلات رودهای-مقعدی، درونزهدانی،[برابرها 60] تخمدانی، معدی-رودهای (اشکالات معدی، روده کوچک، لوزالمعدی و صفراوی)، ادراری، مغزی (گلیبوبلاستوماها[برابرها 61]) و پوستی (کراتوآکاتوماها[برابرها 62] و آدنوماهای چرب[برابرها 63])[48] |
| ژن نشانگان لی-فراوْمنی TP53 | P53 | نقش مستقیم در HRR, BER, NER ایفا میکند و پاسخگوی تخریب DNA است[49] این مسیرهای NHEJ وMMR برای این نشانگان هستند[50] | سارکوماها، سرطانهای سینه، تومورهای مغزی، و کارسینوماهای فوق کلیوی[51] |
| MRE11A | MRE11 | HRR و NHEJ[52] | سینه[53] |
| MUTYH | MUTYH گلوکوزیلاز | BER از یک جفت با 8-oxo-dG[54] | سرطانهای رودهای-مقعدی، دوازدههای، تخمدانی، مثانهای و پوست[55] |
| نشانگان شکست نیژمنگن[برابرها 64] | NBS (NBN) | NHEJ[56] | سرطانهای لمفوئید[57] |
| NTHL1 | NTHL1 | BER برای Tg, FapyG, 5-hC, 5-hU در[58]dsDNA | سرطان روده بزرگ، سرطان درونزهدانی، سرطان دوازدههای، کارسینوما سلول پایهای[برابرها 65][59] |
| RECQL4 | RECQ4 | هلیکاز مشابهاً در HRR فعال است[60] | کارسینومای سلول پایهای، کارسینومای سلول سنگفرشی، کارسینومای بینروپوستی[برابرها 66][61] |
| ژن نشانگان ورنرWRN[برابرها 67] | هلیکاز نشانگان ورنر وابسته به ATP | HRR, NHEJ, مسیر طولانی BER[62] | سارکومای نرم بافتی، رودهای-مقعدی، پوست، تیروئید، لوزالمعده[63] |
| ژنهای خشکپوستی رنگدانهدار XPA, XPB, XPD, XPF, XPG | XPA XPB XPD XPF XPG | رونویسی کامل NER رشتههای رونویسیشدهٔ ژنهای فعال را بهطور رونوشتبردار تعمیر میکنند[64] | سرطان پوست (ملانوما و غیر-ملانوما)[65] |
| ژنهای خشکپوستی رنگدانهدار XPC, XPE (DDB2) | XPC, XPE | ژنومیک جهانیNER، در هر دو DNAی رونویسیشده و رونویسینشده، آسیب میبیند.[66][67] | سرطان پوست (ملانوما و غیر-ملانوما)[68][69] |
| XPV (همچنین پلیمرازH نیز خوانده میشود) | DNAپلیمراز اِتا (Pol η) | سنتز ترا-آسیبش[برابرها 68] (TLS)[70] | سرطان پوست (ملانوما، سلول سنگفرشی و پایهای)[71] |
واژهنامه و برابرها
- Lynch
- hereditary breast-ovarian cancer syndrome, HBOC
- hereditary non-polyposis colon cancer, HNPCC
- adrenocortical carcinoma
- carcinoid tumors
- leiomyosarcoma
- medullary thyroid cancer
- paraganglioma
- pheochromocytoma
- renal cell carcinoma of chromophobe
- hybrid oncocytic
- oncocytoma histology
- sebaceous carcinoma
- sex cord tumors with annular tubules
- allele
- a germline mutation
- Knudson's two hit hypothesis
- sporadic cancers
- homozygous recessive
- autoimmune lymphoproliferative syndrome
- Canale-Smith syndrome
- Beckwith–Wiedemann syndrome
- Birt–Hogg–Dubé syndrome
- Carney syndrome
- Cowden syndrome
- familial chordoma
- dysplastic nevus syndrome with familial melanoma
- familial adenomatous polyposis
- Li-Fraumeni syndrome
- multiple endocrine neoplasia type 1/2
- multiple osteochondromatosis
- Peutz-Jeghers syndrome
- familial prostate cancer
- hereditary leiomyomatosis renal cell cancer , HLRCC
- hereditary papillary renal cell cancer , HPRCC
- hereditary paraganglioma-pheochromocytoma syndrome
- retinoblastoma
- tuberous sclerosis
- von Hippel-Lindau disease
- Wilm's tumor
- ataxia telangiectasia
- Bloom syndrome
- Fanconi anemia
- Rothmund-Thomson syndrome
- Werner's syndrome
- Xeroderma pigmentosum, XP
- aplastic anemia
- myelodysplastic syndrome
- acute myeloid leukemia
- squamous cell carcinomas
- cafe au lait spots
- hypopigmentation
- osteomas
- desmoid tumors
- papillary serous carcinoma of the peritoneum
- microsatellites
- microsatellite instability , MSI
- ataxia telangiectasia
- Bloom syndrome
- endometrial
- glioblastomas
- keratoacanthomas
- sebaceous adenomas
- Nijmegen breakage syndrome
- basal-cell carcinoma
- intraepidermal
- Werner syndrome
- Translesion synthesis
منابع
- Simone Fulda، Heike Allgayer (۲۰۰۹). Hereditary Tumors: From Genes to Clinical Consequences. Weinheim. Wiley-VCH. شابک ۳-۵۲۷-۳۲۰۲۸-۸.
- "Mechanisms of inherited cancer susceptibility". J Zhejiang Univ Sci B. January 2008. doi:10.1631/jzus.B073001. ISSN 2170461 PMC 2170461 Check
|issn=value (help). - "Clinical management of hereditary breast cancer syndromes". J Mammary Gland Biol Neoplasia. April 2011. doi:10.1007/s10911-011-9200-x. ISSN 21360002 PMID 21360002 Check
|issn=value (help). - "Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications". Clin. Genet. ژولای 2009. doi:10.1111/j.1399-0004.2009.01230.x. ISSN 2846640 PMC 2846640 Check
|issn=value (help). Check date values in:|تاریخ=(help) - Bank s, KC; Moline, JJ; Marvin, ML; Newlin, AC; Vogel, KJ (March 2013). "10 rare tumors that warrant a genetics referral". Familial cancer. 12 (1): 1–18. doi:10.1007/s10689-012-9584-9. PMID 23377869.
- Korde, Larissa A.; Gadalla, Shahinaz M. (2017-05-02). "Cancer Risk Assessment for the Primary Care Physician". Primary care. 36 (3): 471–488. doi:10.1016/j.pop.2009.04.006. PMC 2713871. PMID 19616151.
- hereditary diffuse gastric cancer, HDGC
- Anderson, Cindy Lou; Carie A Braun (2007). Pathophysiology: functional alterations in human health. Hagerstwon, MD: Lippincott Williams & Wilkins. ISBN 0-7817-6250-2.
- Lindor NM, Greene MH (July 1998). "The concise handbook of family cancer syndromes. Mayo Familial Cancer Program". J. Natl. Cancer Inst. 90 (14): 1039–71. doi:10.1093/jnci/90.14.1039. PMID 9672254.
- Moldovan GL, D'Andrea AD (2009). "How the fanconi anemia pathway guards the genome". Annu. Rev. Genet. 43: 223–49. doi:10.1146/annurev-genet-102108-134222. PMC 2830711. PMID 19686080.
- Tischkowitz MD, Hodgson SV (January 2003). "Fanconi anaemia". Journal of Medical Genetics. 40 (1): 1–10. doi:10.1136/jmg.40.1.1. PMC 1735271. PMID 12525534.
- Kee Y, D'Andrea AD (November 2012). "Molecular pathogenesis and clinical management of Fanconi anemia". J. Clin. Invest. 122 (11): 3799–806. doi:10.1172/JCI58321. PMC 3484428. PMID 23114602.
- Kottemann MC, Smogorzewska A (January 2013). "Fanconi anaemia and the repair of Watson and Crick DNA crosslinks". Nature. 493 (7432): 356–63. doi:10.1038/nature11863. PMC 3700363. PMID 23325218.
- Su X, Huang J (September 2011). "The Fanconi anemia pathway and DNA interstrand cross-link repair". Protein Cell. 2 (9): 704–11. doi:10.1007/s13238-011-1098-y. PMC 4875268. PMID 21948210.
- Half E, Bercovich D, Rozen P (2009). "Familial adenomatous polyposis". Orphanet J Rare Dis. 4: 22. doi:10.1186/1750-1172-4-22. PMC 2772987. PMID 19822006.
- Galiatsatos P, Foulkes WD (February 2006). "Familial adenomatous polyposis". Am. J. Gastroenterol. 101 (2): 385–98. doi:10.1111/j.1572-0241.2006.00375.x. PMID 16454848.
- Macrae F, du Sart D, Nasioulas S (2009). "Familial adenomatous polyposis". Best Pract Res Clin Gastroenterol. 23 (2): 197–207. doi:10.1016/j.bpg.2009.02.010. PMID 19414146.
- Petrucelli N, Daly MB, Feldman GL (May 2010). "Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2". Genet. Med. 12 (5): 245–59. doi:10.1097/GIM.0b013e3181d38f2f. PMID 20216074.
- Smith EC (2012). "An overview of hereditary breast and ovarian cancer syndrome". J Midwifery Womens Health. 57 (6): 577–84. doi:10.1111/j.1542-2011.2012.00199.x. PMID 23050669.
- Drescher KM, Sharma P, Lynch HT (2010). "Current hypotheses on how microsatellite instability leads to enhanced survival of Lynch Syndrome patients". Clin. Dev. Immunol. 2010: 170432. doi:10.1155/2010/170432. PMC 2901607. PMID 20631828.
- Kunkel TA, Erie DA (2005). "DNA mismatch repair". Annu. Rev. Biochem. 74: 681–710. doi:10.1146/annurev.biochem.74.082803.133243. PMID 15952900.
- Kastrinos F, Syngal S (2011). "Inherited colorectal cancer syndromes". Cancer J. 17 (6): 405–15. doi:10.1097/PPO.0b013e318237e408. PMC 3240819. PMID 22157284.
- Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C (2004). "Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations". JAMA. 292 (8): 943–51. doi:10.1001/jama.292.8.943. PMID 15328326.
- Malkin D (April 2011). "Li-fraumeni syndrome". Genes Cancer. 2 (4): 475–84. doi:10.1177/1947601911413466. PMC 3135649. PMID 21779515.
- Bakry, D (2013). P53 in the Clinic: TP53 Germline Mutations: Genetics of Li–Fraumeni Syndrome. New York: Springer. pp. 167–188. ISBN 978-1-4614-3676-8.
- Birch JM (July 1994). "Familial cancer syndromes and clusters". Br. Med. Bull. 50 (3): 624–39. PMID 7987644.
- Quesnel S, Malkin D (August 1997). "Genetic predisposition to cancer and familial cancer syndromes". Pediatr. Clin. North Am. 44 (4): 791–808. doi:10.1016/s0031-3955(05)70530-7. PMID 9286285.
- Half E, Bercovich D, Rozen P (2009). "Familial adenomatous polyposis". Orphanet J Rare Dis. 4: 22. doi:10.1186/1750-1172-4-22. PMC 2772987. PMID 19822006.
- Sampson JR, Jones N (2009). "MUTYH-associated polyposis". Best Pract Res Clin Gastroenterol. 23 (2): 209–18. doi:10.1016/j.bpg.2009.03.006. PMID 19414147.
- Manfredi M, Vescovi P, Bonanini M, Porter S (March 2004). "Nevoid basal cell carcinoma syndrome: a review of the literature". Int J Oral Maxillofac Surg. 33 (2): 117–24. doi:10.1054/ijom.2003.0435. PMID 15050066.
- Lo Muzio L (2008). "Nevoid basal cell carcinoma syndrome (Gorlin syndrome)". Orphanet J Rare Dis. 3: 32. doi:10.1186/1750-1172-3-32. PMC 2607262. PMID 19032739.
- Richard, S; Gardie, B; Couvé, S; Gad, S (May 30, 2012). "Von Hippel-Lindau: How a rare disease illuminates cancer biology". Seminars in cancer biology. 23 (1): 26–37. doi:10.1016/j.semcancer.2012.05.005. PMID 22659535.
- Henry, Todd; Campell, James; Hawley, Arthur (1969). Todd-Sanford clinical diagnosis by laboratory methods, edited by Israel Davidsohn [and] John Bernard Henry (14th ed.). Philadelphia: Saunders. p. 555. ISBN 0-7216-2921-0.
- Wong WT, n E, Agró Coleman HR, et al. (February 2007). "Genotype–phenotype correlation in von Hippel–Lindau disease with retinal angiomatosis". Archives of ophthalmology. 125 (2): 239–45. doi:10.1001/archopht.125.2.239. PMC 3019103. PMID 17296901. Archived from the original on 12 December 2008. Retrieved 2008-10-22.
- Lehmann AR, McGibbon D, Stefanini M (2011). "Xeroderma pigmentosum". Orphanet J Rare Dis. 6: 70. doi:10.1186/1750-1172-6-70. PMC 3221642. PMID 22044607.
- Niedernhofer LJ, Bohr VA, Sander M, Kraemer KH (2011). "Xeroderma pigmentosum and other diseases of human premature aging and DNA repair: molecules to patients". Mech. Ageing Dev. 132 (6–7): 340–7. doi:10.1016/j.mad.2011.06.004. PMC 3474983. PMID 21708183.
- Bernstein C, Prasad AR, Nfonsam V, Bernstein H. (2013). DNA Damage, DNA Repair and Cancer, New Research Directions in DNA Repair, Prof. Clark Chen (Ed.), شابک ۹۷۸−۹۵۳−۵۱−۱۱۱۴−۶ , InTech, http://www.intechopen.com/books/new-research-directions-in-dna-repair/dna-damage-dna-repair-and-cancer
- Keimling M, Volcic M, Csernok A, Wieland B, Dörk T, Wiesmüller L (2011). "Functional characterization connects individual patient mutations in ataxia telangiectasia mutated (ATM) with dysfunction of specific DNA double-strand break-repair signaling pathways". FASEB J. 25 (11): 3849–60. doi:10.1096/fj.11-185546. PMID 21778326.
- Keimling M, Volcic M, Csernok A, Wieland B, Dörk T, Wiesmüller L (2011). "Functional characterization connects individual patient mutations in ataxia telangiectasia mutated (ATM) with dysfunction of specific DNA double-strand break-repair signaling pathways". FASEB J. 25 (11): 3849–60. doi:10.1096/fj.11-185546. PMID 21778326.
- Thompson LH, Schild D (2002). "Recombinational DNA repair and human disease". Mutat. Res. 509 (1–2): 49–78. doi:10.1016/s0027-5107(02)00224-5. PMID 12427531.
- Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC (2008). "Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair". Proc. Natl. Acad. Sci. U.S.A. 105 (44): 16906–11. doi:10.1073/pnas.0809380105. PMC 2579351. PMID 18971343.
- German J (1969). "Bloom's syndrome. I. Genetical and clinical observations in the first twenty-seven patients". Am. J. Hum. Genet. 21 (2): 196–227. PMC 1706430. PMID 5770175.
- Nagaraju G, Scully R (2007). "Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks". DNA Repair (Amst.). 6 (7): 1018–31. doi:10.1016/j.dnarep.2007.02.020. PMC 2989184. PMID 17379580.
- Lancaster JM, Powell CB, Chen LM, Richardson DL (2015). "Society of Gynecologic Oncology statement on risk assessment for inherited gynecologic cancer predispositions". Gynecol. Oncol. 136 (1): 3–7. doi:10.1016/j.ygyno.2014.09.009. PMID 25238946.
- Thompson LH, Hinz JM (2009). "Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights". Mutat. Res. 668 (1–2): 54–72. doi:10.1016/j.mrfmmm.2009.02.003. PMC 2714807. PMID 19622404.
- Alter BP (2003). "Cancer in Fanconi anemia, 1927-2001". Cancer. 97 (2): 425–40. doi:10.1002/cncr.11046. PMID 12518367.
- Meyer LA, Broaddus RR, Lu KH (2009). "Endometrial cancer and Lynch syndrome: clinical and pathologic considerations". Cancer Control. 16 (1): 14–22. PMC 3693757. PMID 19078925.
- Carethers JM, Stoffel EM (2015). "Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer". World J. Gastroenterol. 21 (31): 9253–61. doi:10.3748/wjg.v21.i31.9253. PMC 4541378. PMID 26309352.
- Kastan MB (2008). "DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture". Mol. Cancer Res. 6 (4): 517–24. doi:10.1158/1541-7786.MCR-08-0020. PMID 18403632.
- Viktorsson K, De Petris L, Lewensohn R (2005). "The role of p53 in treatment responses of lung cancer". Biochem. Biophys. Res. Commun. 331 (3): 868–80. doi:10.1016/j.bbrc.2005.03.192. PMID 15865943.
- Testa JR, Malkin D, Schiffman JD (2013). "Connecting molecular pathways to hereditary cancer risk syndromes". Am Soc Clin Oncol Educ Book. 33: 81–90. doi:10.1200/EdBook_AM.2013.33.81. PMID 23714463.
- Rapp A, Greulich KO (2004). "After double-strand break induction by UV-A, homologous recombination and nonhomologous end joining cooperate at the same DSB if both systems are available". J. Cell Sci. 117 (Pt 21): 4935–45. doi:10.1242/jcs.01355. PMID 15367581.
- Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, Mistrik M, Aittomäki K, Blomqvist C, Heikkilä P, Lukas J, Nevanlinna H, Bartek J (2008). "Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene". Mol Oncol. 2 (4): 296–316. doi:10.1016/j.molonc.2008.09.007. PMID 19383352.
- Markkanen E, Dorn J, Hübscher U (2013). "MUTYH DNA glycosylase: the rationale for removing undamaged bases from the DNA". Front Genet. 4: 18. doi:10.3389/fgene.2013.00018. PMC 3584444. PMID 23450852.
- Patel SG, Ahnen DJ (2012). "Familial colon cancer syndromes: an update of a rapidly evolving field". Curr Gastroenterol Rep. 14 (5): 428–38. doi:10.1007/s11894-012-0280-6. PMC 3448005. PMID 22864806.
- Chrzanowska KH, Gregorek H, Dembowska-Bagińska B, Kalina MA, Digweed M (2012). "Nijmegen breakage syndrome (NBS)". Orphanet J Rare Dis. 7: 13. doi:10.1186/1750-1172-7-13. PMC 3314554. PMID 22373003.
- Chrzanowska KH, Gregorek H, Dembowska-Bagińska B, Kalina MA, Digweed M (2012). "Nijmegen breakage syndrome (NBS)". Orphanet J Rare Dis. 7: 13. doi:10.1186/1750-1172-7-13. PMC 3314554. PMID 22373003.
- Krokan HE, Bjørås M (2013). "Base excision repair". Cold Spring Harb Perspect Biol. 5 (4): a012583. doi:10.1101/cshperspect.a012583. PMC 3683898. PMID 23545420.
- Kuiper RP, Hoogerbrugge N (2015). "NTHL1 defines novel cancer syndrome". Oncotarget. 6 (33): 34069–70. doi:10.18632/oncotarget.5864. PMC 4741436. PMID 26431160.
- Singh DK, Ahn B, Bohr VA (2009). "Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging". Biogerontology. 10 (3): 235–52. doi:10.1007/s10522-008-9205-z. PMC 2713741. PMID 19083132.
- Anbari KK, Ierardi-Curto LA, Silber JS, Asada N, Spinner N, Zackai EH, Belasco J, Morrissette JD, Dormans JP (2000). "Two primary osteosarcomas in a patient with Rothmund-Thomson syndrome". Clin. Orthop. Relat. Res. 378: 213–23. doi:10.1097/00003086-200009000-00032. PMID 10986997.
- Bohr VA (2005). "Deficient DNA repair in the human progeroid disorder, Werner syndrome". Mutat. Res. 577 (1–2): 252–9. doi:10.1016/j.mrfmmm.2005.03.021. PMID 15916783.
- Monnat RJ (2010). "Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology". Semin. Cancer Biol. 20 (5): 329–39. doi:10.1016/j.semcancer.2010.10.002. PMC 3040982. PMID 20934517.
- Menck CF, Munford V (2014). "DNA repair diseases: What do they tell us about cancer and aging?". Genet. Mol. Biol. 37 (1 Suppl): 220–33. doi:10.1590/s1415-47572014000200008. PMC 3983582. PMID 24764756.
- Menck CF, Munford V (2014). "DNA repair diseases: What do they tell us about cancer and aging?". Genet. Mol. Biol. 37 (1 Suppl): 220–33. doi:10.1590/s1415-47572014000200008. PMC 3983582. PMID 24764756.
- Lehmann AR, McGibbon D, Stefanini M (2011). "Xeroderma pigmentosum". Orphanet J Rare Dis. 6: 70. doi:10.1186/1750-1172-6-70. PMC 3221642. PMID 22044607.
- Oh KS, Imoto K, Emmert S, Tamura D, DiGiovanna JJ, Kraemer KH (2011). "Nucleotide excision repair proteins rapidly accumulate but fail to persist in human XP-E (DDB2 mutant) cells". Photochem. Photobiol. 87 (3): 729–33. doi:10.1111/j.1751-1097.2011.00909.x. PMC 3082610. PMID 21388382.
- Lehmann AR, McGibbon D, Stefanini M (2011). "Xeroderma pigmentosum". Orphanet J Rare Dis. 6: 70. doi:10.1186/1750-1172-6-70. PMC 3221642. PMID 22044607.
- Oh KS, Imoto K, Emmert S, Tamura D, DiGiovanna JJ, Kraemer KH (2011). "Nucleotide excision repair proteins rapidly accumulate but fail to persist in human XP-E (DDB2 mutant) cells". Photochem. Photobiol. 87 (3): 729–33. doi:10.1111/j.1751-1097.2011.00909.x. PMC 3082610. PMID 21388382.
- Opletalova K, Bourillon A, Yang W, Pouvelle C, Armier J, Despras E, Ludovic M, Mateus C, Robert C, Kannouche P, Soufir N, Sarasin A (2014). "Correlation of phenotype/genotype in a cohort of 23 xeroderma pigmentosum-variant patients reveals 12 new disease-causing POLH mutations". Hum. Mutat. 35 (1): 117–28. doi:10.1002/humu.22462. PMID 24130121.
- Opletalova K, Bourillon A, Yang W, Pouvelle C, Armier J, Despras E, Ludovic M, Mateus C, Robert C, Kannouche P, Soufir N, Sarasin A (2014). "Correlation of phenotype/genotype in a cohort of 23 xeroderma pigmentosum-variant patients reveals 12 new disease-causing POLH mutations". Hum. Mutat. 35 (1): 117–28. doi:10.1002/humu.22462. PMID 24130121.
